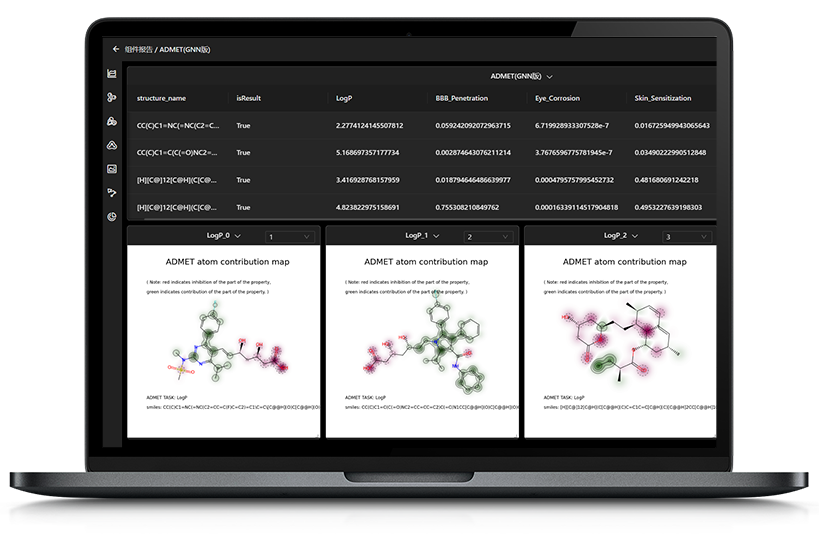
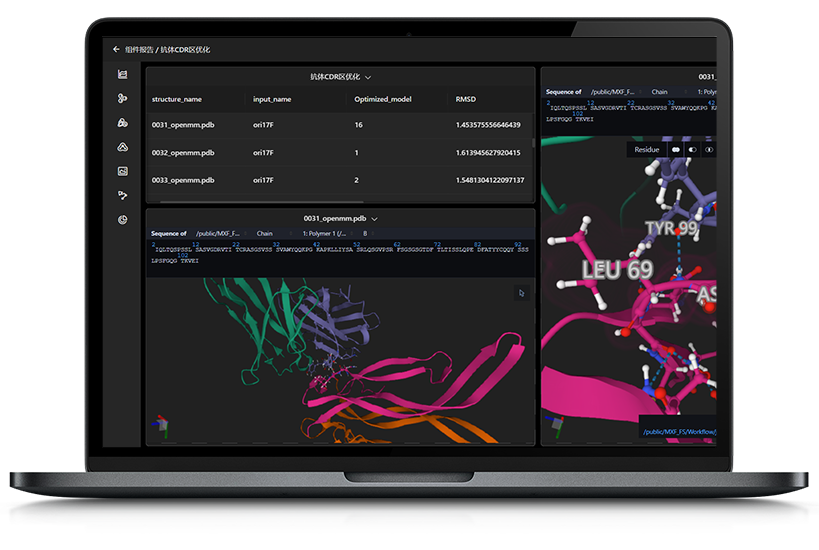
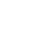▊ 药物研发中结合自由能计算应用现状
药物分子通过对靶蛋白的识别与结合作用,能够调控靶蛋白功能,进而实现治疗疾病的效果。蛋白质的许多关键生理和药理活动是通过与小分子相互作用来实现,比如酶的催化特性是由其与底物的相互作用所体现的。在药物研发中,众多药物和其他生物活性分子的功效实质体现在它们与生物大分子受体的相互协作中,这意味着药物与受体间的结合强度直接影响到药物的活性表现。
结合自由能是评价药物与受体相互作用强度的关键参数之一,它直接关系到药物分子能否有效结合至靶标受体,并进一步影响其生物活性和药理效应。高精度的结合自由能计算一直都是现代基于结构的药物发现工作中关键的组成部分,这类计算通常以统计力学为基础,譬如基于分子动力学模拟的自由能计算。
自由能微扰(FEP)方法是其中最常用的一种计算方法,它能以与实验精度(1kcal/mol)相当的计算精度预测配体和受体之间的相对结合自由能(RBFE)和绝对结合自由能(ABFE),该方法的核心思想就是从一个已知的体系出发,通过一系列微小变化的累积变成另一个体系,其中每一步的变化都是一次分子动力学模拟,最后再利用自由能估算算法从多次的分子动力学模拟中得出自由能的变化。
目前,有效的应用FEP展开药物研发并不简单。即便不考虑初始结构的合理性,仅使用FEP方法进行模拟就需要考虑诸多要素,以RBFE计算为例,需要考虑的要素就包括:拓扑方案、原子间映射、微扰区域、微扰窗口、软核参数等技术要素以及电荷微扰和骨架跃迁等场景要素。这些要素的组合决定了预测的速度、精度、通量和成本。基于FEP方法的开源软件包,诸如PyAutoFEP,往往需要进行精细复杂的配置以及稳固的计算环境构建,然而对于许多化合物pair而言,建立一个既能保持稳定性又能提供可靠计算结果的体系并非易事,这使得它们难以满足工业化应用级别的标准要求。此外,FEP方法存在诸多经验性操作,无疑给那些意图通过定制FEP模拟协议以精确计算各类不同体系结合自由能的研究人员带来了显著的挑战。

▊ 高效精确灵活的解决方案
MaXFlow的Neo-BFE模块,引入了一种名为ATM(Alchemical Transfer Method,炼金术转移法)的创新方法来执行高精度的结合自由能计算。ATM方法提供了一种通过使用坐标变换交换配体位置来计算绝对/相对结合自由能的协议。与常用的FEP方法不同,ATM方法在单个溶剂化盒子中就能完成结合自由能的计算(可避免配体净电荷变化的影响),不仅减少了计算量,还极大简化了计算流程。此外,由于该方法无需像FEP那样通过开、关闭配体与受体之间的静电和非静电相互作用项,来实现配体与受体之间的解耦,进而模拟计算出结合自由能,因此,该方法能兼容任何势能函数(如可极化,量子力学以及机器学习势),同时还能避免FEP执行过程中的那些经验性较强的技术和场景因素带来的问题。
▊ 打造结合自由能计算的“灰盒”工具
Neo-BFE模块的设计目标是将结合自由能计算作为一个“灰盒” ,即允许用户根据计算任务的实际需求对过程进行精细化的控制,同时也可以直接采取它提供的“最佳实践”默认值以减少个人干预和实现自动化。因此无论是专家或非专家用户均可使用Neo-BFE模块来进行稳健、高效、准确的结合自由能计算。
▊ 计算性能
精度方面,Neo-BFE支持机器学习力场和哈密顿副本交换增强采样的算法,可在兼顾计算精度的同时做到充分采样;速度方面,经过优化后的分子动力学模拟积分器- Langevin Middle Integrator 会使得模拟更稳定,在添加氢质量再分配情况下,可将默认时间步长从2fs增加到4fs而不损失稳定性,使得模拟所需要的GPU时间减少近两倍,在推荐的参数下一次结合自由能计算能够在5-10小时内完成,基于MXF的云计算平台可以调用海量资源一天完成成百上千次的自由能计算。易用性方面,因为不需要像FEP那样定义复杂的原子映射关系,微扰区域,软核或软键势等经验性较强的参数,所以和常规动力学类似,简单的设置便能得到高精度的计算结果,同时,MaXFlow平台实现了计算流程的极大自动化,可轻易的进行高通量计算,灵活分配计算资源。
目前,Neo-BFE中的ATM方法已达到与实验精度相当的结合自由能预测值(1 kcal/mol)。在已发表的论文中,研究人员通过选择Schrödinger论文中发表的数据集进行测试(包括8 个不同蛋白质系统中的330个配体对),并将ATM方法与其他常见方法(例如 FEP+、Amber 和 pmx)进行比较,验证它们在相对结合自由能预测方面的性能[1]。结果表明,ATM 在Pearson相关性方面与更复杂的自由能扰动 (FEP) 方法的性能相匹配甚至超越。尽管其平均误差略高,但考虑到ATM方法在实现上的简便性、灵活性以及在势函数通用性方面的优势,这一方法在药物发现中有着巨大的应用前景和价值。
表1:自由能方法的性能比较,8个被测蛋白的Pearson相关性(r)和平均绝对误差(MAE) (单位kcal/mol)

▊ MaXFlow新一代分子模拟与人工智能平台
在MaXFlow找到该模块,通过拖拽组件的形式搭建如下工作流,选择要计算的结构,设置相应的参数即可运行该工作流。本次案例使用的结构是PDB ID:4HBV。

表中展示了计算得到的自由能变化(ΔG)的值,该值为-7.7 Kcal/mol表示有利于驱动化学反应的进行。标准差为0.4 Kcal/mol,相较于ΔG的绝对值来说较小,所以可以认为计算出的ΔG=-7.7 Kcal/mol是比较准确和可靠的。使用的约束类型是FloatBottom。采用哈密顿副本交换增强采样,每个副本采样时间为1ns,采样间隔为10ps,因此每个副本采样数为100个。分析采用未分箱加权直方图分析方法(UWHAM)。
表2: ATM结合自由能计算结果

下图展示的是两条炼金术路径的扰动能量概率密度分布图。从图中可以看出,在扰动能量比较小的时候,采样很充分,重叠也很好。但是当扰动逐渐增大时,出现了不够重叠的概率密度分布,这预示着1ns的采样时间可能不太够,需要更加充分的采样,以获得更良好的自由能估计。


▊ 总结
在药物研发中,结合自由能计算对于评估药物分子与受体蛋白的相互作用至关重要。传统自由能微扰(FEP)方法虽能精确预测结合自由能,但实施复杂且受限于多种技术因素。MaXFlow的Neo-BFE模块创新地引入了ATM方法,通过坐标变换简化并加速结合自由能计算,克服了FEP方法的部分局限性,提供更高兼容性、稳定性和计算效率。Neo-BFE设计为“灰盒”工具,既方便专家精细化调控,也适合非专家用户快速准确地进行大规模计算。实验证明,ATM方法在结合自由能预测上达到与FEP相当的精度,展现出强大的应用潜力,尤其在药物发现领域。
[1] Sabanés Zariquiey F,Pérez A,Majewski M,et al.Validation of the Alchemical Transfer Method for the Estimation of Relative Binding Affinities of Molecular Series.[J].J Chem Inf Model,2023,63,8:2438-2444.
扫码立即体验「分子模拟与人工智能平台」