酶是驱动生物体内化学反应的引擎,是维持生命所不可或缺的物质。然而与一般的催化剂不同,酶作为蛋白质极易受到外界条件影响,从而变性失去催化活性。因此,自然界中的酶通常只能在特定条件下发挥作用,难以适应工业生产中的复杂和多变环境。酶工程可以解决这些由于野生型蛋白自身性质而导致无法工业化的问题。通过酶工程,我们可能设计获得催化效率更高、稳定性更好、选择性更佳的新型酶。
目前,分子动力学模拟(MD)是计算机辅助酶设计的常用工具,该方法利用牛顿运动定律,对目标酶及其周围环境的原子进行时间步长为飞秒级别的运动模拟,从而得到酶分子在一定时间内的构象变化和能量变化。MD可以揭示酶催化反应的微观机理,分析酶与底物和抑制剂之间的分子相互作用,预测酶结构和功能在不同条件下的变化,帮助理解酶如何催化反应,以及如何修改酶以提高其活性和特异性。

图1. 酶工程方法简史。在过去的几十年里,为设计酶而采取的方法(黑色文本)随着对酶结构、功能和动力学及其相互联系的理解(绿色文本)的进展而发展。
(来源:https://doi.org/10.1093/protein/gzac015)
分子动力学在酶设计中的要点是选择合适的MD模拟方案,包括力场、溶剂模型、边界条件、温度和压力控制以及模拟长度。不同的方案可能对模拟的准确性和效率有不同的影响,并且可能需要不同水平的计算资源。因此,有必要针对不同的酶系统比较和验证不同的方案,并选择最符合研究目标的方案。

图2. 刀豆球蛋白A(ConA)/三甘露糖苷(3MAN)复合物和一分子水。使用不同的水模型(TIP3P和TIP5P),能量计算的结果也不同
(来源:Elisa Fadda)
创腾科技的MaXFlow平台提供了多样的力场、多种水溶剂模型和丰富的模拟参数设置选项以及可视化页面,适用于各类型体系的模拟。

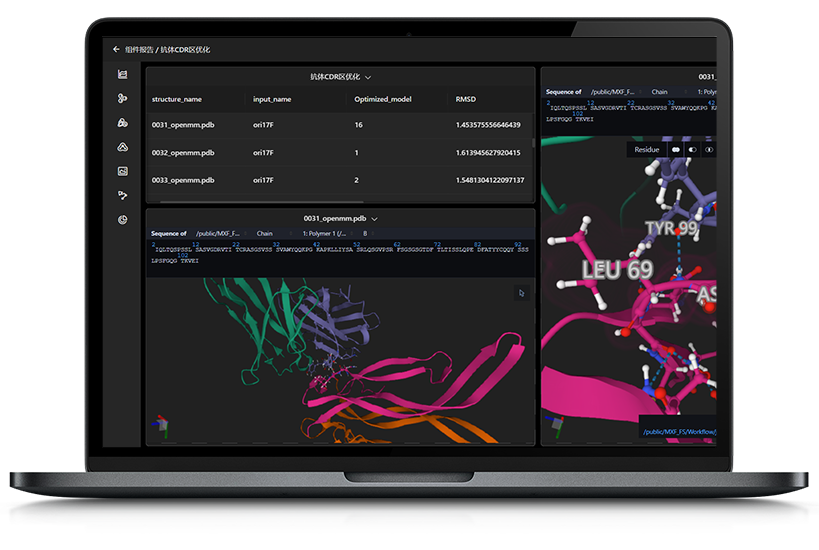
图3. MaXFlow酶蛋白结构构建

图4. MaXFlow快速构建用于分子动力学模拟的周期性体系
分子动力学模拟的结果可以提供关于酶结构和动力学的很多信息,例如构象变化、氢键网络、静电相互作用、结合自由能和活化能垒。然而,这些信息并不总是可靠或与实验数据一致。因此,有必要检查模拟的质量,例如收敛性、稳定性和可重复性,并将模拟结果与现有的实验数据或其他计算方法进行比较。
创腾科技的MaXFlow平台集成了丰富的轨迹分析功能,可以实现分子动力学模拟和分析的一体化流程。MaXFlow平台便捷易用的工作流引擎可以自动计算和绘制各种物理量随时间变化的曲线(例如RMSD、RMSF、温度、压力、能量、回旋半径、二级结构、溶剂可及表面积等),以及各种物理量之间的相关性(例如氢键和盐桥的距离、键角、二面角、寿命等)。

图5. MaXFlow高效便捷的分子动力学模拟工作流

图6. MaXFlow丰富的轨迹分析功能展示(部分)。从左到右从上到下:RMSD分析、RMSF分析、盐桥分析、二级结构分析、溶剂化能计算
单独使用MD可能不足以或有效地为特定反应或底物设计酶。因此,将MD与其他计算方法或实验技术结合起来有助于对酶设计的全面理解,例如分子对接、自由能计算、定向进化或突变。朱玉山课题组使用MD模拟结合MM/PBSA和MM/GBSA方法,评估头孢拉定水解酶的活性设计,计算了头孢拉定水解酶设计变体的结合自由能和活化能垒。模拟结果与实验数据的Km和Kcat/Km进行了比较,发现MM/PBSA和MM/GBSA方法都能根据预测的结合自由能很好地对变体的活性进行排序。这些方法可以帮助识别可能增强酶活性或特异性的潜在突变或修饰,并评估它们对酶结构和功能的影响。这些计算均可在MaXFlow平台实现。


图7. 模拟结果∆Gbind与实验数据的Km和Kcat/Km的对比(来源:RSC Adv., 2019, 9, 13868)


图8. MaXFlow中利用MM-PB/GBSA工具进行结合自由能计算和丙氨酸扫描
酶设计中使用计算模拟的一个挑战是结构数据库的不完善或缺乏活性构象。深度学习的方法能够从零开始生成新颖的蛋白质序列和框架。这些创新工具的广泛应用将为酶设计开辟新的可能性。

图9. Baker和Houk课题组使用深度学习模型从头设计荧光素酶。设计模型(蓝色)和 AlphaFold2 预测模型(灰色)在主干和侧链均具有较高的一致性。
(来源:https://doi.org/10.1038/s41467-020-19594-z)